
Pediatric Polycystic Kidney Disease: Practice Essentials, Etiology, Epidemiology

The 3 basic processes involved in renal cyst formation and progressive enlargement are as follows
[4, 5] :
-
Abnormalities in tubular extracellular matrix and/or function
Tubular cell hyperplasia
This may be mediated by factors that control cell proliferation (eg, epidermal growth factor, transforming growth factor-α), dysregulation of apoptosis, or the balance between the 2.
Tubular fluid secretion
The solid tumor cell nests produced by the cell hyperplasia described above are transformed into fluid-filled cysts by the secretion of fluid by the tubular cells associated with efferent tubular obstruction or slow or absent afferent flow. This accounts for the fluid within the cysts of kidneys in patients with autosomal dominant polycystic kidney disease, 70% of which have no afferent or efferent tubular connections.
Abnormalities in tubular extracellular matrix and/or function
These are thought to be responsible for amplifying tubular cell hyperplasia and tubular fluid secretion. Interstitial inflammation and fibrosis are responsible for progression in all forms of polycystic kidney disease.
Autosomal recessive polycystic kidney disease
In 1994, the autosomal recessive polycystic kidney disease gene (PKDHD1) was localized to the short arm of chromosome 6.
[6] Fibrocystin/polyductin, a protein encoded by PKDHD1, is expressed on the cilia of renal and bile duct epithelial cells and is thought to be crucial in maintaining the normal tubular architecture of renal tubules and bile ducts. However, the precise function of this protein has yet to be completely studied or understood. The protein strengthens the theory that the primary defect in autosomal recessive polycystic kidney disease is linked to ciliary dysfunction.
[7]
Autosomal recessive polycystic kidney disease is characterized by nonobstructive, bilateral, symmetrical dilatation and elongation of 10-90% of the renal collecting ducts, focally accounting for a wide variability of renal dysfunction. As the number of ducts involved increases, the kidneys enlarge. However, at autopsy, the reniform shape is maintained, because the abnormality is in the collecting ducts and the cysts are usually minute (< 3 mm). In older patients, cysts as large as 1 cm may be seen. (See the images below.)
Excretory urogram shows minimal bilateral tubular changes caused by a mild form of autosomal recessive polycystic kidney disease (ARPKD).
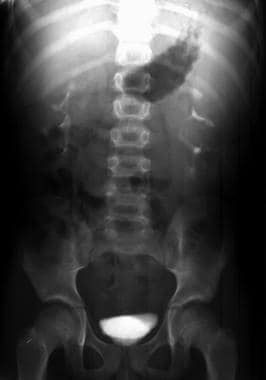
Excretory urogram shows enlarged kidneys with bilateral distortion of the collecting system (spider-legs configuration). These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
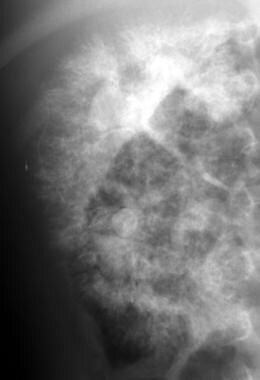
Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
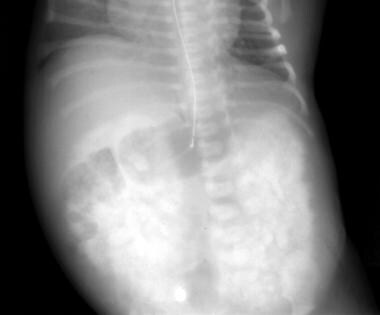
Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
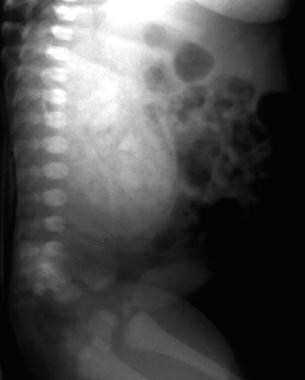
Excretory urogram shows the typical mottled (spongelike) contrast enhancement pattern in autosomal recessive polycystic kidney disease (ARPKD).
At autopsy, gross examination of a kidney in patients with autosomal recessive polycystic kidney disease reveals multiple minute cystic spaces throughout the capsular surfaces. Cut sections of the kidney show that these cystic structures are subcapsular extensions of radially oriented cylindrical or fusiform ectatic spaces, with poor corticomedullary differentiation due to the extension of the elongated and dilated collecting ducts from the medulla to the cortex.
All patients with autosomal recessive polycystic kidney disease have congenital hepatic fibrosis (CHF), which may have a more severe clinical manifestation than the renal disease. The CHF results from malformation of the developing ductal plate. The liver biopsy findings reveal enlarged, fibrotic portal tracts and hyperplastic, dilated, and dysgenetic biliary ducts with normal hepatocytes. The ductules can show true cystic changes, and, when the changes are macroscopic, autosomal recessive polycystic kidney disease can be indistinguishable from Caroli disease. The portal hypertension secondary to the CHF can be clinically debilitating, with splenomegaly, varices, and GI hemorrhage.
[8]
The results from one study noted that characteristics of CHF are similar in both autosomal dominant and autosomal recessive polycystic kidney diseases.
[9]
In a study designed to better understand the complications of autosomal recessive polycystic kidney disease, researchers at the NIH analyzed clinical, molecular and imaging data from 73 patients (ages 1-56 years old, average 12.7) with mutations in PKHD1 and kidney and liver involvement. The findings identified platelet count as the best predictor of the severity of portal hypertension, which has early onset but is underdiagnosed in patients with autosomal recessive polycystic kidney disease.
[10]
A study that included 304 patients with autosomal recessive polycystic kidney disease found that biallelic null variants in PKHD1 are associated with severe disease and a poor prognosis. For example, variants that affect amino acids 2625-4074 of fibrocystin are linked to worse hepatic outcomes.
[11]
Autosomal dominant polycystic kidney disease
The genes responsible for autosomal dominant polycystic kidney disease were localized to the short arm of chromosome 16 (PKD1) in 85% of cases and the long arm of chromosome 4 (PKD2) in most of the remaining cases. The proteins encoded by PKD1 and PKD2 are polycystin 1 and polycystin 2, respectively. These proteins are expressed in the developing kidney, and their functions overlap considerably.
The dysfunction of these proteins is thought to be pathogenetically responsible for the manifestations of autosomal dominant polycystic kidney disease, primarily by renal ciliary dysfunction. Whether a third gene accounts for a small number of unlinked families is uncertain. Homozygous or compound heterozygous genotypes have been thought to be lethal in utero. Individuals heterozygous for PKD1 and PKD2 mutations usually survive to adulthood but have more severe renal disease.
Autosomal dominant polycystic kidney disease differs from autosomal recessive polycystic kidney disease in that cysts associated with autosomal dominant polycystic kidney disease develop anywhere along the nephron. Upon clinical presentation, kidneys are usually enlarged, with numerous large, round nodules on the external surface of the kidney, causing the loss of its original reniform shape, which is different from kidneys in patients with autosomal recessive polycystic kidney disease.
Cysts of varying sizes containing pale fluid or blood are randomly distributed throughout the parenchyma and involve any segment along the nephron. The cysts have thickened basement membranes with pericystic interstitial fibrosis, and their epithelium maintains active secretion and reabsorption. It has been hypothesized that patients with an associated marked epithelial hyperplasia may have a higher rate of malignant transformation than does the general population.
link
: A Comprehensive Guide")





