
Less common phenotypes of myelin oligodendrocyte glycoprotein antibody-related diseases in children deserve more attention

We retrospectively analyzed 236 children with MOGAD, of which 196 cases (83.1%) had typical phenotypes, including ADEM, ON, TM, and NMOSD, and 40 cases (16.9%) had less common clinical phenotypes, including 16 cases of (6.8%) cortical encephalitis, 13 of (5.5%) MNOS, 7 of (3.0%) cerebral leukodystrophy-like phenotype, 3 of (1.3%) isolated seizures without any findings on MRI, and 3 of (1.3%) aseptic meningitis. Among these, two cases of cortical encephalitis overlapped with MNOS (Fig. 1).
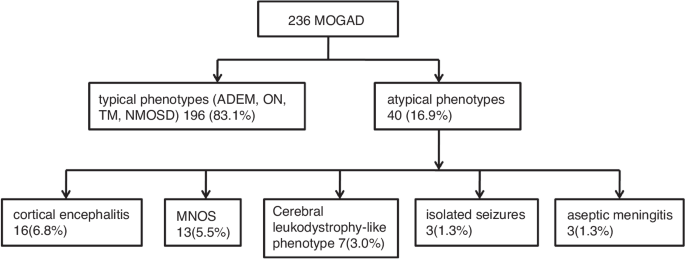
Two hundred and thirty-six children with MOGAD were analyzed, of which 40 cases had less common clinical phenotypes, including 16 cases of cortical encephalitis, 13 of MNOS, 7 of cerebral leukodystrophy-like phenotype, 3 of isolated seizures without any findings on MRI, and 3 of aseptic meningitis.
Cortical encephalitis
The clinical features, imaging characteristics, and treatment details of 16 children with cortical encephalitis phenotype are shown in Table 1. A total of 18 episodes of illness were observed in these children, with two patients experiencing two episodes. Seizures, the most commonly observed clinical symptoms, were seen in 16 episodes (88.9%). There were 12 (75%) bilateral tonic–clonic seizures of focal origin and 9(52.3%) status epilepticus. Other symptoms were headache in 13 (72.2%), fever in 11 (61.1%), dyskinesia in 7 (38.8%), hemiparesis in 5 (27.8%), speech disorder in 5 (27.8%), and cognitive deficits in 5 (27.8%). MOG antibodies were positive in 12 episodes, which were negative or undetectable at the time of the first episode in six cases. The CSF leukocytes fluctuated from 4 to 136 ×10^6/L, with 11 cases (61.1%) having a mononuclear cell count of >15 ×10^6/L. Six episodes (33.3%) had increased CSF protein levels (0.48–2.81 g/L). Cranial MRI of these children showed cortical hyperintensities on T2-weighted fluid-attenuated inversion recovery sequences. Six cases (33.3%) had sulcus or periaqueductal enhancement; 11 cases (61.1%) had unilateral cortical abnormalities and seven (38.9%) had bilateral cortical abnormalities. The frontal lobe was the most common site to be affected (66.7%), followed by the temporal lobe (38.9%), parietal lobe (33.3%), and occipital lobe (22.2%).
As shown in Table 2, 15 children (93.8%) were misdiagnosed at the disease onset, of which 10 (66.6%) were misdiagnosed with viral encephalitis, 3 (20.0%) with epilepsy, 1 (6.7%) with purulent meningitis, and 1 (6.7%) with stroke. At the initial diagnosis, these 15 cases received anti-infective therapy and five of them received antiepileptic therapy, with no improvement after 9 (interquartile range, IQR: 5–20) days of treatment. Nine cases received immunotherapy and showed significant improvement after 2 (IQR: 1–3) days of therapy. Three children without immunotherapy were discharged with the diagnosis of viral encephalitis; however, during follow-up, two of them developed ADEM and one developed MNOS, which later improved after receiving immunotherapy. Two cases were initially hospitalized with the diagnosis of epilepsy, and their symptoms improved after antiepileptic therapy before discharge. At a follow-up of 48 (IQR: 36–55) months, 11 cases (68.8%) relapsed after 1–11 months following the first episode of cortical encephalitis, with a total of 19 relapses. 4 (44.4%) of the 9 children treated with immunotherapy after the first attack had a relapse after 17.5 (IQR: 9.5–23.3) months ; the other 7 children who were not treated with immunotherapy had a relapse 1 (IQR: 1–2) month after the first attack, with eight episodes of (42.1%) ON, six of (31.6%) ADEM, two of (10. 5%) MNOS, and three of (15.8%) cortical encephalitis.
MNOS
As shown in Fig. 2, 13 children with MNOS had a total of 30 acute episodes. At first attack, the most common clinical symptoms were seizures (92.3%), followed by mental-behavioral abnormalities (84.6%), language disorders (84.6%), motor disorders (84.6%), cognitive disorders (76.9%), consciousness disorders (76.9%), sleep disorders (61.5%), and visual disorders (38.5%). At the time of first onset, MNOS was diagnosed in seven (53.8%), MOGAD in four (30.8%), and NMDAR in two (15.4%) cases. Moreover, 10 of the 30 acute episodes who had symptoms of both MOGAD and anti-NMDAR encephalitis were positive for both blood MOG antibodies and CSF anti-NMDAR, seven (23.3%) showed as anti-NMDAR encephalitis symptoms only (such as abnormal psychiatric behavior, sleep disorder and cognitive dysfunction), and nine (30%) showed as MOGAD symptoms only(such as visual impairment, headache and consciousness disturbance). Furthermore, 13 children had supratentorial lesions on MRI (100%), with involvement of the subcortical white matter in 11 (84.6%), cortical gray matter in 8 (61.5%), basal ganglia in 7 (53.8%), and thalamus in 5 (38.5%) episodes. Moreover, infratentorial lesions were observed in eight cases (61.5%), including five (38.5%) in the cerebellum and four (30.8%) in the brainstem.

The most common clinical symptoms were seizures (12/13), followed by abnormal (psychiatric) behavior (11/13), speech disorders (11/13), motor dysfunction (11/13), cognitive dysfunction (10/13), disturbance of consciousness (10/13), sleep disorders (8/13).
As shown in Table 3, with a median follow-up of 43 (IQR: 27–49) months, 11 cases (84.6%) relapsed. On average, the first relapse occurred 4 (IQR: 3–9) months after the first disease onset, the second relapse occurred at 17.5 (IQR: 12.3–27.3) months after the first onset, and the third relapse occurred at 46 months after the first onset in one child. Seven children received first-line immunotherapy (intravenous methylprednisolone and/or intravenous immunoglobulins) after the first diagnosis of MNOS, and five children (71.4%) relapsed by the last follow-up, with an annual relapse rate of 0.5 (IQR: 0.33–1). After the first relapse, 10 children received maintenance immunotherapy (five received mycophenolate mofetil, five received rituximab, one received regular intravenous immunoglobulin, and one received azathioprine), and three children (30%) relapsed by the last follow-up, with an annual relapse rate of 0.5 (IQR: 0.4–0.5), which was lower than the annual relapse rate of children not receiving immunotherapy (median 2.4, IQR: 2–2.9).
Cerebral leukodystrophy-like phenotype
Seven cases of cerebral leukodystrophy-like phenotype were noted, with a median age of onset of 5.3 (IQR: 4.1–6.5) years. Four (57.1%) of the seven children had motor disorders (three had hemiplegia and one had ataxia), three had consciousness disturbance (42.9%), three had cognitive disorders (42.9%), three had seizures (42.9%), three had headaches (42.9%), two had a fever (28.6%), two had visual disturbances (28.6%), two had speech disturbances (28.6%), two had personality changes (28.6%), and one had facial palsy (14.3%). The early symptoms of six children met the diagnostic criteria for ADEM. All seven children were positive for MOG antibodies, with titers ranging from 1:10 to 1:100. The cranial MRIs of all seven children showed cerebral leukodystrophy foci, involving subcortical white matter in seven (100.0%), deep white matter in five (71.4%), basal ganglia in five (71.4%), optic nerves in three (42.9%), thalamus in two (28.6%), brainstem in two (28.6%), and cerebellum in two (28.6%) cases. Figure 3 shows MRI brain images of three of these cases.
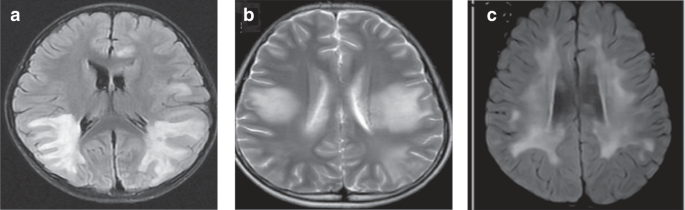
a Paraventricular symmetric white matter lesions. b Large supratentorial symmetric cerebral white matter lesions. c Diffuse subcortical white matter changes.
These children received immunotherapy at a median duration of 15 (IVIg+HIMP, IQR: 11–22) days after disease onset, with clinical improvement and a median Expanded Disability Status Scale score of 3 (IQR: 1.5–4.5) and 1 (IQR: 0–1) in the acute phase and at the time of discharge, respectively. One case was lost to follow-up, and the remaining six children were followed for a median duration of 27 (IQR: 12–42) months, with three children (50.0%) developing relapses. By the last follow-up, two children (33.3%) had visual impairment, one (16.7%) had epilepsy, and one (16.7%) had cognitive impairment.
Isolated seizures without any findings on MRI
Among three cases of isolated seizures without any findings on MRI, the only clinical symptom was epileptic seizures, except for one case that presented with transient dizziness and headache that resolved quickly. Two cases (66.7%) had focal motor seizures and one (33.3%) had focal motor seizures and focal secondary bilateral tonic–clonic seizures. Two cases had normal electroencephalograms, and one case had focal spike-like slow waves in the sleep phase. Three children had normal cranial MRIs. Epilepsy was diagnosed at the time of the first seizure in two cases and viral encephalitis at the time of the first seizure in one case. In one case, a serum MOG antibody of 1:10 was measured at the initial onset. Serum MOG antibody was not tested in two cases at initial seizure onset, and their MOG antibody titer was measured to be 1:100 at the time of the reoccurrence of the demyelinating event.
The follow-up lasted for 20–44 months ; two cases had demyelinating events within 2 months after discharge, both presenting as ADEM, which subsided following immunotherapy. Two cases were persistently seropositive for MOG antibodies, and MOG antibodies turned negative in one case.
Aseptic meningitis
All three children with aseptic meningitis had a fever and showed signs of meningeal irritation. Two cases had a fever for more than 2 weeks and one case had a fever for 5 days, with a mean duration of fever of 15.7 days. At the disease onset, the white blood cell counts were increased (13.38–27.45 × 10^9/L), with neutrophils predominating, with a serum CRP level of >8 mg/L in one case and normal serum CRP level in the rest. The serum PCT level was normal in all three cases before and after immunotherapy. CSF leukocytes were increased in all cases, and the CSF protein level was elevated by 1.31 g/L in one case. After immunotherapy, the CSF leukocyte and protein level were significantly decreased in two cases, and one case did not recheck the CSF. The serum MOG antibody titers of all three children ranged from 1:100 to 1:320. Two children were initially diagnosed with purulent meningitis and treated with anti-infective therapy for 19 and 23 days, respectively; no improvement was observed. Their symptoms significantly improved after receiving immunotherapy for 2 days. One child was initially diagnosed with viral meningoencephalitis and treated with antiviral therapy for 8 days with the addition of intravenous immunoglobulin, which showed marked improvement.
At 19–57 months of follow-up, one child was lost to follow-up and the remaining two had complete clinical remission without recurrence. The MRI lesion completely resolved in one case.
Table 4 shows the clinical information of the above five phenotypes.
link
: A Comprehensive Guide")





